
Bayesian MCMC methods have become incredibly popular in recent times as they allow the implementation of arbitrarily complex models for various statistical inference problems. In molecular phylogenetics, MCMC has been used to estimate species phylogenies, species divergence times, and species delimitation under the multi-species coalescent, among many other applications. However, many key concepts and issues of MCMC appear to be arcane to the average scientist. The purpose of this tutorial is thus to open the MCMC black box, and provide a step-by-step guide on how to write an MCMC program and explore the main concepts and issues involved.
This tutorial focuses on writing a Bayesian MCMC algorithm to estimate the molecular distance between two species (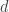
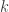
(
A biologist’s guide to Bayesian phylogenetic analysis
Nascimento FF, dos Reis M and Yang Z (2017) Nature Ecology and Evolution, 1:1446–1454.
The tutorial is divided into 4 parts:
Part 1: Introduction
Part 2: Markov chain Monte Carlo (MCMC)
Part 3: Efficiency of the MCMC chain
Part 4: Diagnosing the MCMC chain
The complete R script can be downloaded from github.com/thednainus/Bayesian_tutorial.

Pingback: Part 1: Introduction | The DNA in Us